The genomic DNA sequence of the New World cottontail rabbit (S. floridanus) contains 228,444,470 nucleotide bases (or 2.85 Gb; "Gb" notation is short for "gigabases"), occupying 2.7 GiB of disk space (uncompressed). This dataset is available on NCBI as assembly mSylFlo1.10. It is smaller than human genomic DNA, which has 3,092,479,710 bases (or 3.09 Gb; assembly GRCh38.p11 a.k.a. hg38), sits uncompressed on disk at 3.1 GiB.
How hard would it be to find a protein homolog with the size of 748 residues (or 2,244 bases) in human against this ocean of 2.85 Gb? Such size represents 0.000026% of the available search space. A letter-sized paper can probably fit 7,314 bases. At a print speed of 20 pages per minute (single-sided, 0.7-inch margin, in Comic Sans 10pt), ~26 hours later we could have printed a stack of papers standing tall at 6.2 feet (comprising 31,234 total pages from 62 reams) containing all nucleotides from this bunny.
We could potentially look for that 2,244 bases this way. At a sobering 75 characters/second of human scanning speed, that would take 846 hours of uninterrupted work. For 21-weeks of a 40-hour work week (assuming a single reader), that probably does not sound too bad, except that biological complexity compounds the difficulty.
Among litany of rules governing the existence of a gene, here is a small list:
- A gene must start with the
ATGcodon[1] (encoding for methionine residue). - A gene must stop with one of the stop codons.
That seems tractable.
Assuming a random distribution (err...) of nucleotides, we could probably mark 3,569,445 (or 3.57 million) ATG codons, highlight those stretches of DNA that are then terminated by a stop codon, cut those pieces out, and paste them somewhere.
The list of rules continues as follows:
- Those stretches of DNA must be in multiple of 3. Now we have to count more diligently.
- Must be of a certain length to encode for meaningfully functional protein. This probably helps with the extent of search space.
- Probably enclosed by DNA cis-regulatory elements. This increases the complexity of such search.
- Might have splice sites as a gene may be composed of several exons spaced by several introns. This sounds annoying to identify.
At this point, I was convinced that this is not a job for mere mortals. Several decades ago, we tricked sand and stones into doing math and nowadays these central processing units (CPUs) can calculate with their speed clocked at billions of computations per second, maybe they can do this better than us?
In late June 2025, I took a challenge to identify the TAP1 homolog in S. floridanus. The web tblastn (peptide query on nucleotide database) on the NCBI portal offered little help.
The bunny's genome was mostly unannotated at the time of investigation, and the lack of transcriptomic data from multiple tissues complicates the searching process.
Functionally, this would be an exercise of using grep in the most unhinged way.
With limited training in bioinformatics and years of seeking refuge within terminal, I started swimming in this ocean.
Warning
This is not meant to be a tutorial. Code snippets here do not include the whole process and written for brevity.
Strategy I - BLAST+ Suite
Running BLAST locally involves two sequential steps: (a) building a database from .fasta files with makeblastdb tool and (b) running queries against this database with any of the BLAST tools (i.e. tblastn tool).
These two steps are straightforward (assuming the familiarity of running command line applications) and would only take seconds to complete.
# completes in 14.3s
# produces 8 files with prefix 'database.*'
makeblastdb -in GCA_949820135.1_mSylFlo1.10_genomic.fna -dbtype nucl -out database
# completes in 38.7s
# saves the result as 'blast_results.txt' plaintext file
tblastn -query huTAP1.fasta -db database -out blast_results.txt \
-evalue 1e-5 -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore sseq" \
-max_target_seqs 50I picked up BLAST+ suite in April 2020, early during the SARS-CoV-2 outbreak. A few weeks after the pandemic declaration followed by the sequencing frenzy for outbreak surveillance motivated me to attempt at tracking SARS-CoV-2's evolutionary path during its transmission in human populations for tell-tale signs of further adaptation and immune evasion. This was an easy BLAST task; the viral genome is small (30 Kb +ssRNA) and the rules are simpler.
However, I could not reliably find the full-length TAP1 homolog with just makeblastdb and tblastn as it is not intron/exon/CDS-aware.
The returned matches were short (avg: ~60 residues) when we inspected the results written to blast_results.txt file.
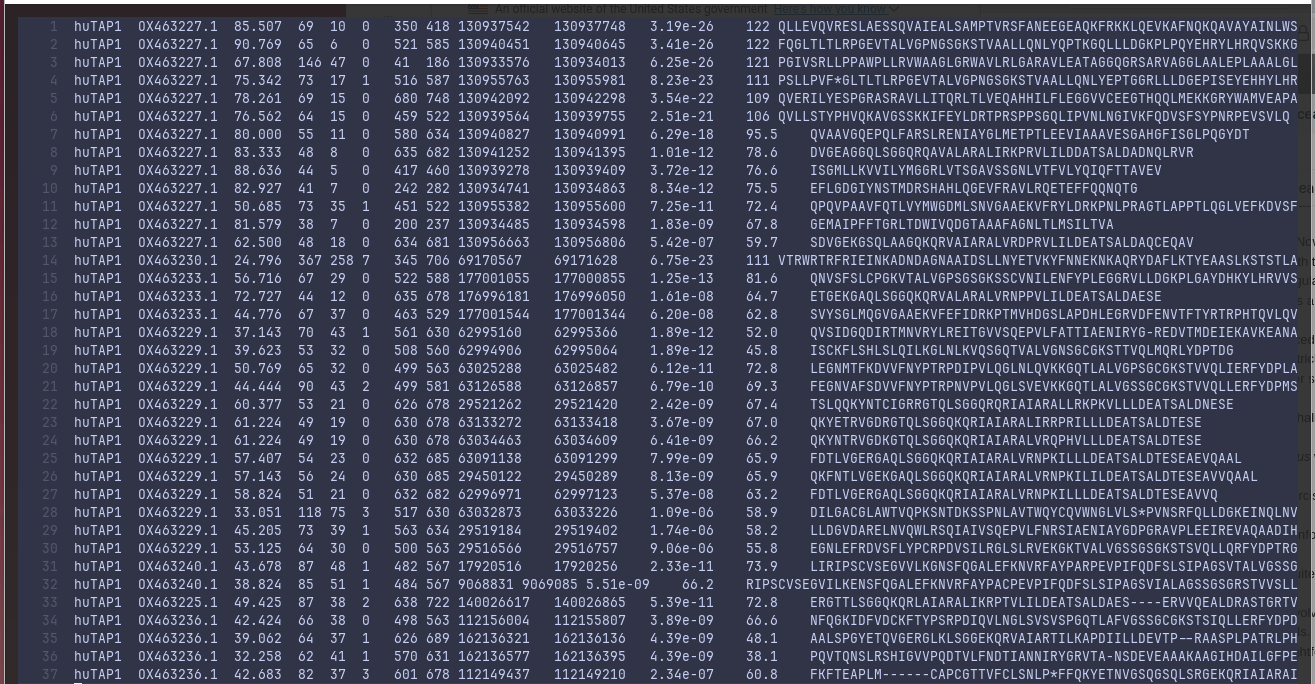
Notwithstanding that, the majority of top hits came from OX463227.1 (the ID for chromosome 5).
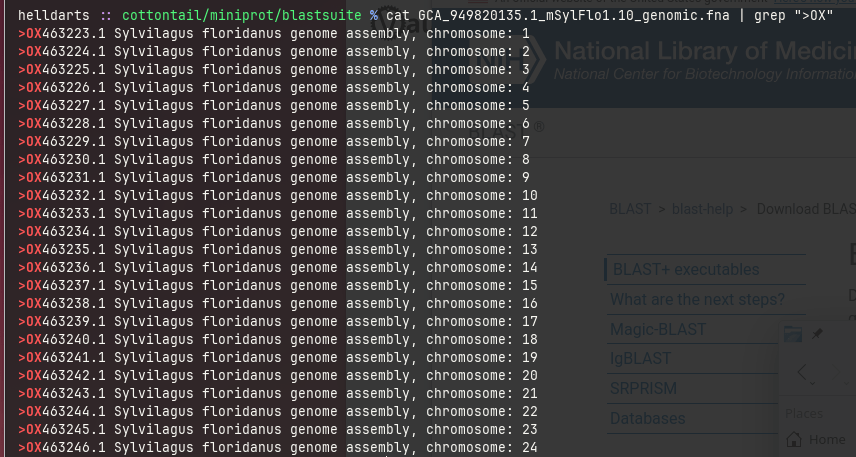
2.7 GiB complete genomic DNA is probably resource-intensive to traverse even with specialized tools, but narrowing down to chromosome 5 alone could speed up discovery. Despite the lack of success, the BLAST+ suite took a cumulative 53 seconds traversing the whole genome that is similar to reading 53,743,179 bases per second, equivalent to reading 7,348 pages in a blink.
Humans could never.
Strategy II - Gene Prediction with Augustus
A web search looking for a gene prediction tool landed me on Augustus GitHub repo.
The codebase contains a Dockerfile, signaling to me some degree of architectural engineering for modern deployments[2].
The docker build step produced a 3.69 GB container image labeled as augustus:latest now available for use.
I am sure there is a better way to work with this container as my way was awkward.
# clone the repo and build the container
# building the image took me 416s (~7 minutes)
git clone https://github.com/Gaius-Augustus/Augustus.git
cd Augustus
docker build -t augustus .
# drop into the container's shell
# with current directory mounted at /work inside the container
docker run -it --rm \
-v "$(pwd)":/work:rw \
augustus:latest /bin/bash
# begin predicting inside the container
# took 58'20" to complete
# identified 1,335 potential genes
augustus --species=human --gff3=on --uniqueGeneId=true --codingseq=on --cds=on \
/work/chr5.fasta > /work/augustus-predictions-chr5.gff3... where a .fasta file containing the entire chromosome 5 (chr5.fasta) was first extracted by using seqkit (shenwei356/seqkit on GitHub), a utility program written in Go[3].
seqkit grep -p "OX463227.1" GCA_949820135.1_mSylFlo1.10_genomic.fna > chr5.fastaAugustus is trained to predict for genes of the entire input genome ab initio fashion, that is, unassisted[4] during run owing to the datasets it had previously seen.
It is aware of exons/introns, terminal UTRs, alternative transcripts, and rules that are species-specific.
Unfortunately, Augustus was not trained on rabbits or related species, in which I deferred to passing human to the --species= parameter[5].
Because it needs to meticulously find for every single possible gene, the runtime was crawling.
Note
Initially when I tried Augustus on the whole S. floridanus genome, it took approximately 17 hours to identify 24,016 genes, or 23 genes/minute. On the other hand, chromosome 5 only contains 193,479,110 bases, representing 6.8% of the entire genome of S. floridanus and that took 58 minutes to complete.
The output is augustus-predictions-chr5.gff3 (GFF3 = general feature format, version 3; in plaintext) with a filesize of 4.3 MiB.
A simple text search using the most conserved TAP1 residues did land a hit.
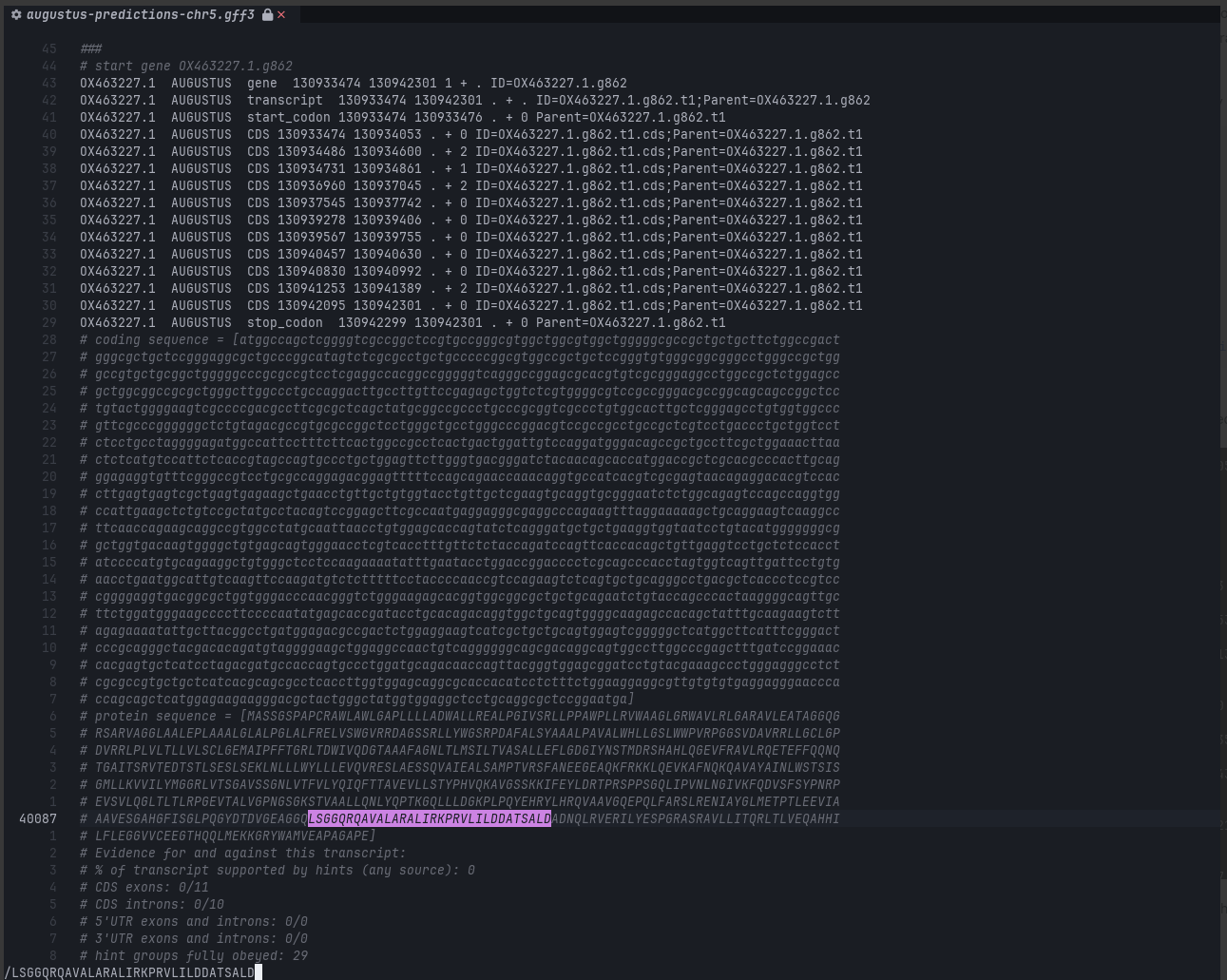
I could have called it a day at this step, except, I wanted to produce an amino acid .fasta (or .faa to contrast with .fna for nucleic acid) file.
This would yield two benefits: a cleaner way to record putative proteins identified from chr5.fasta, and additionally, BLAST-able with blastp.
Gemini 2.5 Flash co-authored a simple program written in Go to extract polypeptide sequences from this augustus-predictions-chr5.gff3 result.
# parse to extract polypeptide sequences
go run gff3_parser.go augustus-predictions-chr5.gff3 > proteins_chr5.fasta
# prepare a BLAST database
makeblastdb -in proteins_chr5.fasta -dbtype prot -out proteins_chr5_db
# run blastp
blastp -query huTAP1.fasta -db proteins_chr5_db -out blast_results.txt \
-evalue 1e-5 -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore sseq" \
-max_target_seqs 50 Lucky! 74.5% identity for the 748 queried residues with 145 mismatches and 3 gaps.
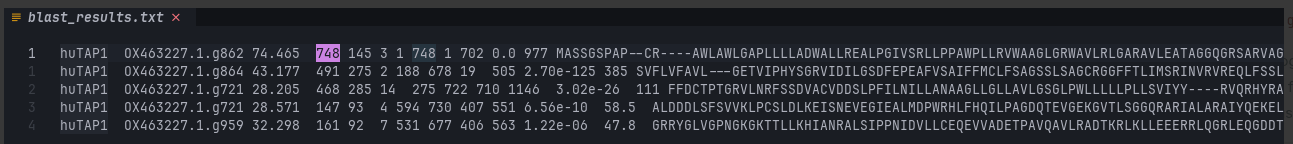
To compare, here are the polypeptide sequences for TAP1 from H. sapiens and S. floridanus:
>S_floridanus|702aa
MASSGSPAPCRAWLAWLGAPLLLLADWALLREALPGIVSRLLPPAWPLLRVWAAGLGRWA
VLRLGARAVLEATAGGQGRSARVAGGLAALEPLAAALGLALPGLALFRELVSWGVRRDAG
SSRLLYWGSRPDAFALSYAAALPAVALWHLLGSLWWPVRPGGSVDAVRRLLGCLGPDVRR
LPLVLTLLVLSCLGEMAIPFFTGRLTDWIVQDGTAAAFAGNLTLMSILTVASALLEFLGD
GIYNSTMDRSHAHLQGEVFRAVLRQETEFFQQNQTGAITSRVTEDTSTLSESLSEKLNLL
LWYLLLEVQVRESLAESSQVAIEALSAMPTVRSFANEEGEAQKFRKKLQEVKAFNQKQAV
AYAINLWSTSISGMLLKVVILYMGGRLVTSGAVSSGNLVTFVLYQIQFTTAVEVLLSTYP
HVQKAVGSSKKIFEYLDRTPRSPPSGQLIPVNLNGIVKFQDVSFSYPNRPEVSVLQGLTL
TLRPGEVTALVGPNGSGKSTVAALLQNLYQPTKGQLLLDGKPLPQYEHRYLHRQVAAVGQ
EPQLFARSLRENIAYGLMETPTLEEVIAAAVESGAHGFISGLPQGYDTDVGEAGGQLSGG
QRQAVALARALIRKPRVLILDDATSALDADNQLRVERILYESPGRASRAVLLITQRLTLV
EQAHHILFLEGGVVCEEGTHQQLMEKKGRYWAMVEAPAGAPE
>H_sapiens|748aa
MASSRCPAPRGCRCLPGASLAWLGTVLLLLADWVLLRTALPRIFSLLVPTALPLLRVWAV
GLSRWAVLWLGACGVLRATVGSKSENAGAQGWLAALKPLAAALGLALPGLALFRELISWG
APGSADSTRLLHWGSHPTAFVVSYAAALPAAALWHKLGSLWVPGGQGGSGNPVRRLLGCL
GSETRRLSLFLVLVVLSSLGEMAIPFFTGRLTDWILQDGSADTFTRNLTLMSILTIASAV
LEFVGDGIYNNTMGHVHSHLQGEVFGAVLRQETEFFQQNQTGNIMSRVTEDTSTLSDSLS
ENLSLFLWYLVRGLCLLGIMLWGSVSLTMVTLITLPLLFLLPKKVGKWYQLLEVQVRESL
AKSSQVAIEALSAMPTVRSFANEEGEAQKFREKLQEIKTLNQKEAVAYAVNSWTTSISGM
LLKVGILYIGGQLVTSGAVSSGNLVTFVLYQMQFTQAVEVLLSIYPRVQKAVGSSEKIFE
YLDRTPRCPPSGLLTPLHLEGLVQFQDVSFAYPNRPDVLVLQGLTFTLRPGEVTALVGPN
GSGKSTVAALLQNLYQPTGGQLLLDGKPLPQYEHRYLHRQVAAVGQEPQVFGRSLQENIA
YGLTQKPTMEEITAAAVKSGAHSFISGLPQGYDTEVDEAGSQLSGGQRQAVALARALIRK
PCVLILDDATSALDANSQLQVEQLLYESPERYSRSVLLITQHLSLVEQADHILFLEGGAI
REGGTHQQLMEKKGCYWAMVQAPADAPEClustal Omega alignment came out showing a substantial stretch of residues is missing and I was informed that that region is highly conserved.

My victory celebration was premature.
Strategy III - miniprot
The Augustus strategy bore fruit, but I decided to run another test to hunt for the missing chunk and hoping for something that could run faster. I asked Claude Sonnet 4 for advice and it replied with several promising leads: Exonerate, Helixer, BRAKER, and miniprot. Of these 4 options, I leaned towards miniprot due to its relatively young age (published in 2023) and I recognized its author (Heng Li, lh3 on GitHub) from my previous work building a deep sequencing analysis pipeline for influenza virus. Heng Li (of Dana-Farber Cancer Institute) and collaborators are prolific bioinformaticians whose creations massively contributed to the field of genomic analysis.

Acquiring miniprot is straightforward.
Pre-compiled binary for linux is available, or we can compile from source ourselves.
# clone the repo and compile
git clone https://github.com/lh3/miniprot
cd miniprot && make
# index chromosome 5
# took 3.3" to complete
miniprot -t8 -d genome_chr5.mpi chr5.fasta
# query chromosome 5
# took 0.2" to complete
miniprot -Iut8 --gff --trans genome_chr5.mpi huTAP1.fasta > aln_tap1_chr5.gff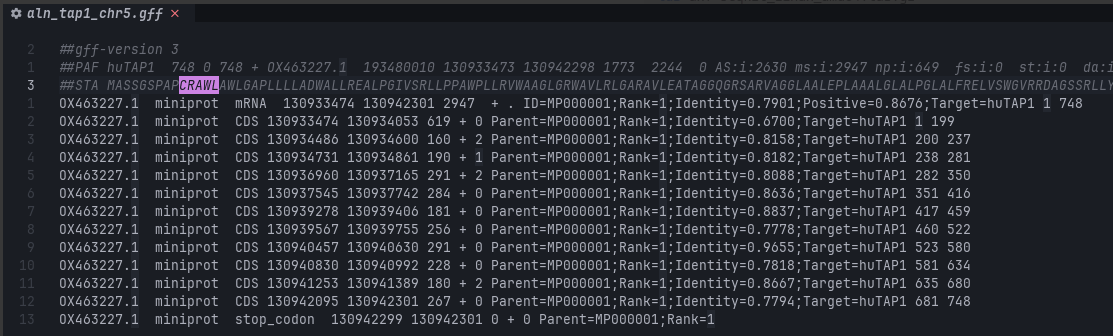
Unlike blastp where multiple hits were returned, miniprot only returned 1 hit in this case.
>S_floridanus|742aa
MASSGSPAPCRAWLAWLGAPLLLLADWALLREALPGIVSRLLPPAWPLLRVWAAGLGRWA
VLRLGARAVLEATAGGQGRSARVAGGLAALEPLAAALGLALPGLALFRELVSWGVRRDAG
SSRLLYWGSRPDAFALSYAAALPAVALWHLLGSLWWPVRPGGSVDAVRRLLGCLGPDVRR
LPLVLTLLVLSCLGEMAIPFFTGRLTDWIVQDGTAAAFAGNLTLMSILTVASALLEFLGD
GIYNSTMDRSHAHLQGEVFRAVLRQETEFFQQNQTGAITSRVTEDTSTLSESLSEKLNLL
LWYLVRGLCLMGLMLWESLSLTMVTLLTLPLLFLLPKKLGKWYELLEVQVRESLAESSQV
AIEALSAMPTVRSFANEEGEAQKFRKKLQEVKAFNQKQAVAYAINLWSTSISGMLLKVVI
LYMGGRLVTSGAVSSGNLVTFVLYQIQFTTAVEVLLSTYPHVQKAVGSSKKIFEYLDRTP
RSPPSGQLIPVNLNGIVKFQDVSFSYPNRPEVSVLQGLTLTLRPGEVTALVGPNGSGKST
VAALLQNLYQPTKGQLLLDGKPLPQYEHRYLHRQVAAVGQEPQLFARSLRENIAYGLMET
PTLEEVIAAAVESGAHGFISGLPQGYDTDVGEAGGQLSGGQRQAVALARALIRKPRVLIL
DDATSALDADNQLRVERILYESPGRASRAVLLITQRLTLVEQAHHILFLEGGVVCEEGTH
QQLMEKKGRYWAMVEAPAGAPE742 residues? Potentially an improvement from our previous strategy (702 residues), and maybe we have that missing chunk?

Perfect.
I think we got him.
All these works (index and query) with miniprot took a sum of 3.5 long seconds.
If you are a photon, you could have travelled approximately 1,049,273,603 meters within 3.5 seconds at the speed of light.
While I wished I could go faster, it had been several days banging my head to get all these to work. It is time for some closing words as I gathered rivulets of life lessons from this exercise.
Epilogue
Taking jabs at our reading speed was probably unfair. Our pattern recognition is still unmatched; gigabytes of data collected by our eyes are streamed toward brain for processing in real-time with milliseconds latency.
We just suck at reading.
Under the right circumstances, well-engineered computer programs can identify pieces of information buried deep within stacks of data at a terrifying accuracy and unfathomable speed. For genome annotation primarily in non-model eukaryotic organisms (i.e., anything beyond human, mouse, fly, yeast, worm, rat, zebrafish, etc), being able to extract homologs is essential to understanding evolution at a granular detail. Extracting the protein-coding genes is one part of the story, as there are cis- and trans-regulatory elements that dictate the level of gene expressions. Less than two weeks ago, Google DeepMind released AlphaGenome, another Google-led breakthrough in attempting to untangle and to model the cis-regulatory elements and effect of variants. Regulatory elements are difficult to understand not only because they are harder to control and to account for, but the fact that multiple layers of control can exist, complicating experimentation and observation.
This exercise on searching for gene was a discovery, in that, while I lack formal bioinformatic training (my doctoral training was in viral immunology), I could supplement the missing pieces with years of using linux as my primary operating system[6] (and being comfortable with terminal operations), some foundational knowledge in writing computer programs, both tied together by curiosity in problem-solving. That said, my theoretical foundational knowledge in genomics and statistics would discourage me from calling myself a bioinformatician. I am no more than a monkey with a hammer seeing problems as nails I could smack with extreme prejudice.
After all, writing computer programs involves passing values around most of the time.
Transporter associated with antigen processing 1 (TAP1)
TAP1 protein forms the heterodimeric TAP complex together with its partner, TAP2. Its deficiency is compatible with life[7], but that life would be royally unpleasant, plagued with recurrent microbial infections with disease phenotypes characteristic of severe immunodeficiency. The TAP heterodimer is essential for the activity of major histocompatibility complex (MHC) class I, crucial for the eventual loading short peptides on the said class I molecules. Without it, nucleated cells that are infected cannot effectively signal to surveilling immune cells that it needs to be executed to prevent the spread of infection to neighboring cells.
Note
I avoided going deeper into the detailed mechanism of TAP, MHC-I, CD8, TCR, GrzB et al. because this essay would be too long for a short coffee break between assay incubations.
The TAP heterodimer is embedded in the endoplasmic reticulum (ER) membrane. As with most membrane-embedded proteins, crystallographic analysis to reveal their quaternary structures is often met with difficulties. That said, the structure of TAP complex was solved with cryo-EM at a somewhat blurry resolution of 6.5 – 4.0 Å[8].
This was an interesting difficult target for protein large language models (LLMs) to predict the folding.
It was among my first targets for testing the Boltz-2 model.
During the .a3m generation step with MMseqs2 through colabfold_batch, I noticed that the sequence coverage was particularly awful with the first half of the H. sapiens TAP1 protein (TAP2 is also similar).
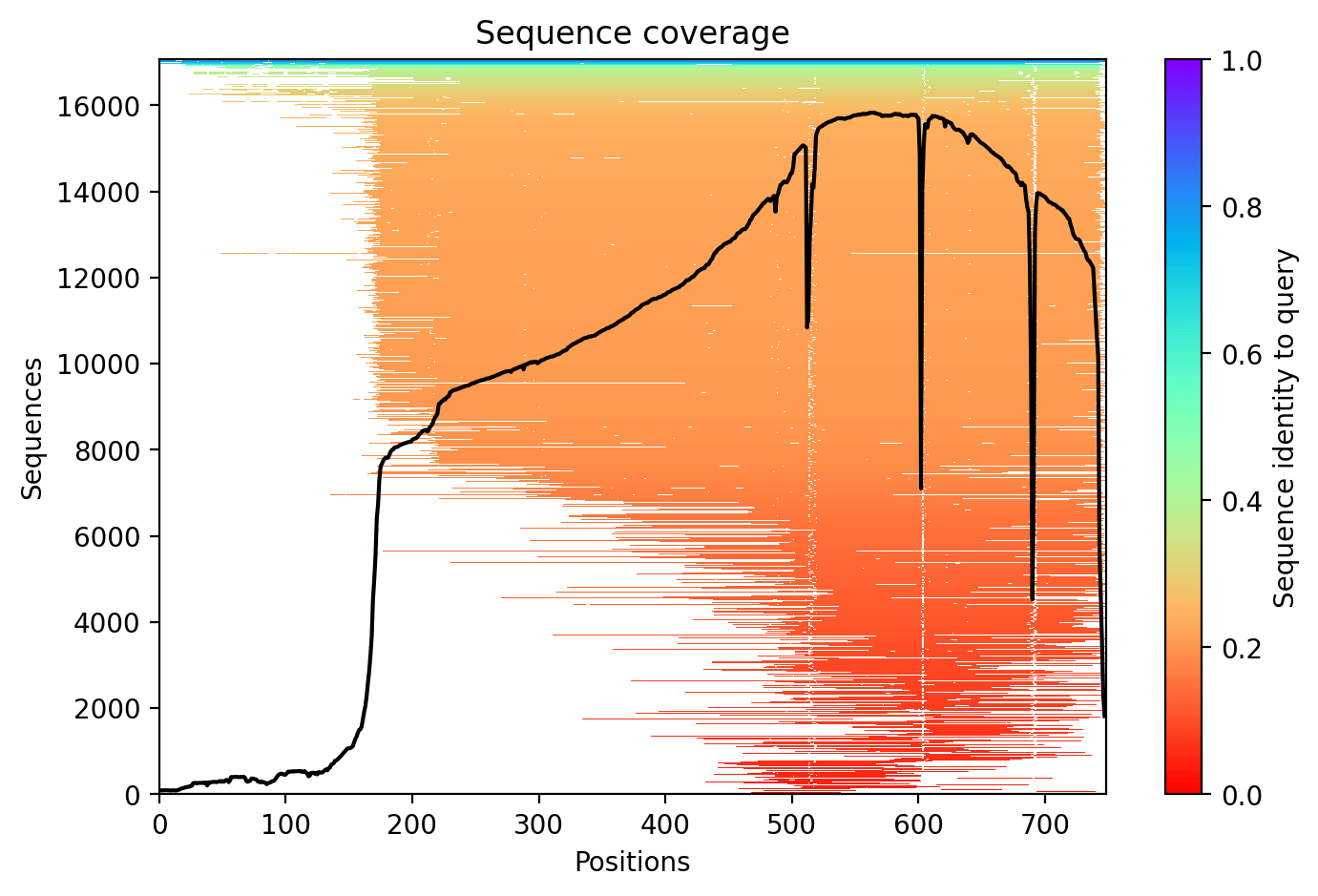
Interestingly, the published cryo-EM data by Michael Oldham and colleagues (eLife 2016) also indicated that their solved structure for TAP1 spanned between residue 173 to 742 due to the diffulty of obtaining higher resolution in the N-terminus, similarly recapitulating the lack of information up to the 172nd residue in the sequence coverage map shown above. This inspired me to think about collecting sequences of TAP1 across the animal kingdom and predict their structure with protein language models for eventual RMSD calculations, and as a fun little excuse to teach myself how to fish for genes from unannotated genomes.
LLM Disclaimer
LLMs did not write this essay. However, LLMs did help with calculations and to clarify certain concepts. In particular, Gemini 2.5 Flash helped me write Go programs and Sonnet 4 helped me with searching the internet for tips and tricks.
All sentences were hand-crafted.
Ribosomes aren't perfect. They can miss (i.e. leaky scanning). See Dmitry E. Andreev et al. (Genome Biol. 2022) "Non-AUG translation initiation in mammals". ↩︎
a fancy way of saying "no need to hunt for libraries to successfully compile this program". ↩︎
I am advocating for more bioinformatic tools written in Go for a true cross-platform single-binary deployments (yes, I am aware Java exists but c'mon... it is 2025). Moreover, the built-in multithreading primitive with goroutine enables for effortless concurrency. It was insane that I could execute a Go program that counted all bases in S. floridanus genome in 22 seconds, all 2.85 billion of them! Humans could never! ↩︎
Often for producing the most optimal predictions, transcriptomic data from multiple tissues can help to accurately identify genes. ↩︎
Note that Augustus can be re-trained, see Retraining AUGUSTUS or this copy of retraining.html on its GitHub repo. ↩︎
I use Arch, btw. On a similar note, this essay was written with the Helix editor. ↩︎
Refer to Luc Van Kaer et al. (Cell 1992) "TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4−8+ T cells" ↩︎
Atomic resolution of sub-3.5 Å enables a high-fidelity reconstruction of protein structure. Note that this is not a criticism of the study that solved this structure. ↩︎
Published on 2025/Jul/05 // Aizan Fahri
aixnr[at]outlook[dot]my